
Découverte de 42 locus significatifs à l’échelle du génome associés à la dyslexie
Une étude réalisée au sein de l’Université d’Édimbourg et publiée dans Nature genetics le 20 octobre 2022 a analysé l’ensemble des génomes de plus d’un million d’individus de partout dans le monde.
Résumé
La lecture et l’écriture sont des compétences vitales essentielles, mais environ un enfant sur dix souffre de dyslexie, qui peut persister à l’âge adulte. Des études familiales sur la dyslexie suggèrent une héritabilité allant jusqu’à 70 %, mais peu de marqueurs génétiques convaincants ont été trouvés. Ici, nous avons réalisé une étude d’association à l’échelle du génome de 51 800 adultes déclarant un diagnostic de dyslexie et 1 087 070 témoins et identifié 42 locus significatifs indépendants à l’échelle du génome : 15 dans les gènes liés à la capacité cognitive/au niveau d’instruction, et 27 nouveaux et potentiellement plus spécifiques à dyslexie. Nous avons validé 23 locus (13 nouveaux) dans des cohortes indépendantes d’ascendance chinoise et européenne. L’étiologie génétique de la dyslexie était similaire entre les sexes, et une covariance génétique avec de nombreux traits a été trouvée, y compris l’ambidextrie, mais pas les mesures neuroanatomiques des circuits liés au langage.
Principal
La capacité de lire est cruciale pour la réussite scolaire et l’accès à l’emploi, à l’information et aux services de santé et sociaux, et est liée au statut socioéconomique atteint 1 . La dyslexie est un trouble neurodéveloppemental caractérisé par de graves difficultés de lecture, présent chez 5 à 17,5 % de la population, selon les critères diagnostiques 2 , 3 . Il implique souvent un traitement phonologique altéré (le décodage des unités sonores, ou phonèmes, dans les mots) et coexiste fréquemment avec des troubles psychiatriques et d’autres troubles du développement 4 , en particulier le trouble déficitaire de l’attention avec hyperactivité (TDAH) 5 , 6 et les troubles de la parole et du langage 7 , 8. La dyslexie peut représenter l’extrême inférieur d’un continuum de capacité de lecture, un trait multifactoriel complexe avec des estimations d’héritabilité allant de 40 % à 80 % 9 , 10 . L’identification des facteurs de risque génétiques aide non seulement à mieux comprendre les mécanismes biologiques, mais peut également étendre les capacités de diagnostic, facilitant l’identification précoce des personnes sujettes à la dyslexie et aux troubles concomitants pour un soutien spécifique.
Les enquêtes précédentes portant sur l’ensemble du génome de la dyslexie se limitaient à des analyses de liaison des familles affectées 11 ou à des études d’association modestes ( n < 2 300 cas) d’enfants et d’adolescents diagnostiqués 12 . Les gènes candidats des études de liaison montrent une réplication incohérente et les études d’association à l’échelle du génome (GWAS) n’ont pas trouvé d’associations significatives, bien que LOC388780 et VEPH1 aient été pris en charge dans les tests basés sur les gènes 12. Des cohortes plus importantes sont essentielles pour augmenter la sensibilité afin de détecter de nouvelles associations génétiques de faible effet. Ici, nous présentons le plus grand GWAS de dyslexie à ce jour, avec 51 800 adultes déclarant eux-mêmes un diagnostic de dyslexie et 1 087 070 témoins, qui sont tous des participants à la recherche avec la société de génétique personnelle 23andMe, Inc. Nous validons nos découvertes d’association dans des cohortes indépendantes, fournissons des annotations de variants significatifs (principalement des polymorphismes mononucléotidiques (SNP)) et des gènes causaux potentiels, et estimations de l’héritabilité basée sur les SNP. Enfin, nous étudions les corrélations génétiques avec la lecture et les compétences connexes, la santé, les mesures socio-économiques et psychiatriques, et évaluons les preuves des gènes candidats de la dyslexie précédemment impliqués dans nos résultats puissants.
Résultats
Associations à l’échelle du génome
L’ensemble de données complet comprenait 51 800 participants (21 513 hommes, 30 287 femmes) ayant répondu « oui » à la question « Avez-vous reçu un diagnostic de dyslexie ? » (cas) et 1 087 070 (446 054 hommes, 641 016 femmes) participants ayant répondu « non » (témoins). Les participants étaient âgés de 18 ans ou plus (l’âge moyen des cas et des témoins était de 49,6 ans (ET 16,2) et 51,7 ans (ET 16,6), respectivement). Nous avons identifié 42 locus associés significatifs indépendants à l’échelle du génome ( P < 5 × 10 -8 ) et 64 loci avec une signification suggestive ( P < 1 × 10 -6 ) (Fig. 1 et tableau supplémentaire 1 ). L’inflation génomique était modérée ( λ GC = 1,18) et compatible avec la polygénicité (voir graphique Q – Q, Extended Data Fig. 1 ). Nous avons également réalisé des GWAS spécifiques au sexe et des GWAS spécifiques à l’âge (plus jeune ou plus âgé que 55 ans) car la prévalence de la dyslexie était plus élevée chez nos jeunes (5,34 % chez les 20 à 30 ans) que chez les plus âgés (3,23 % chez les 80 à 80 ans). participants de 90 ans). Ces analyses de sous-échantillons ont montré une grande cohérence avec le GWAS principal (de l’échantillon complet). La corrélation génétique estimée par la régression du score de déséquilibre de liaison (LD) (LDSC) était de 0,91 (intervalles de confiance (IC) à 95 % : 0,86–0,96 ; P = 8,26 × 10 -253 ) chez les hommes et les femmes, et de 0,97 (IC à 95 % : 0,91 –1,02 ; P = 2,32 × 10 −268 ) entre les adultes plus jeunes et plus âgés.
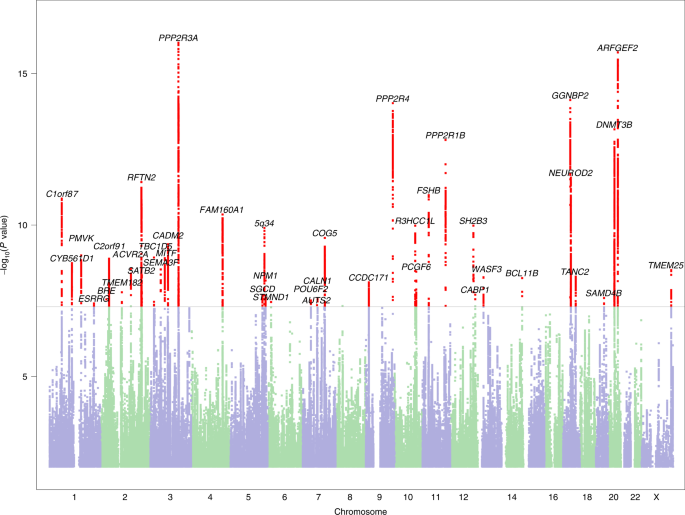
Sur les 17 variants significatifs à l’échelle du génome dans le GWAS féminin (Extended Data Fig. 2 ), tous sauf quatre (rs61190714, rs4387605, rs12031924 et rs57892111) étaient significatifs dans le GWAS principal et, sur ces quatre, trois étaient en LD avec un SNP qui se rapprochait de la signification ( P < 3,3 × 10 −7 ou moins) dans l’analyse principale. Le SNP intergénique rs57892111 (situé entre TFAP2B et PKHD1 sur le chromosome 6p) ne figurait pas parmi les SNP significatifs ou suggestifs de l’analyse principale et peut donc représenter une variante spécifique à la femelle. Il n’y a aucune preuve de GWAS existant que ce SNP est associé à un autre trait humain. Parmi les six variantes significatives à l’échelle du génome du GWAS mâle (Extended Data Fig. 3), tous étaient significatifs dans la principale GWAS.
Dans le GWAS principal, toutes les variantes significatives étaient autosomiques, à l’exception de rs5904158 à Xq27.3 (pour les parcelles d’association régionales, voir la Fig. 1 supplémentaire ). Un total de 17 variantes d’indice étaient en LD élevé avec des SNP associés publiés (significatifs à l’échelle du génome) dans le catalogue NHGRI GWAS 13 (15 étaient associés à des traits cognitifs/éducatifs ; tableaux supplémentaires 1 et 2 ). Ainsi, un total de 27 locus associés n’ont montré aucune preuve d’associations publiées à l’échelle du génome avec des traits censés se chevaucher avec la dyslexie (par exemple, le niveau d’instruction, la capacité cognitive) et ont été considérés comme nouveaux (tableau 1 ).Tableau 1 Nouvelles associations de SNP avec la dyslexie, y compris les résultats basés sur les gènes, le statut eQTL, l’expression dans le cerveau et la validation dans trois cohortes indépendantes (GenLang Consortium, CRS et NeuroDys)
Sur 38 locus associés (les 4 restants ont été marqués par des indels non disponibles dans les cohortes de validation), 3 (rs13082684, rs34349354 et rs11393101) étaient significatifs à un niveau corrigé de Bonferroni ( P < 0,05/38) dans la méta-analyse GWAS du consortium GenLang de capacité de lecture ( n = 33 959) et d’orthographe ( n = 18 514) 14 . À P < 0,05, 18 étaient associés dans GenLang, 3 dans le cas-témoin NeuroDys GWAS 12 ( n = 2 274 cas) et 5 dans l’étude de lecture chinoise (CRS) sur la précision et la fluidité de la lecture ( n = 2 270 ; Note supplémentaire ) (Tableau 1 et Tableaux supplémentaires 3 –6).
Les tests basés sur les gènes ont identifié 173 gènes significativement associés (tableau supplémentaire 7 ), mais aucune voie biologique significativement enrichie (tableau supplémentaire 8 ). Nous avons estimé que l’héritabilité de la dyslexie basée sur le SNP sur l’échelle de responsabilité LDSC était h 2 SNP = 0,152 (erreur standard = 0,006) en utilisant la prévalence de l’échantillon 23andMe de 5 %, et h 2 SNP = 0,189 (erreur standard = 0,008) en utilisant un 10 % de prévalence de la dyslexie, qui est plus typique de la population générale 2 , 3 .
Cartographie fine et annotations fonctionnelles
Dans l’ensemble de variantes crédibles (tableau supplémentaire 1 ), les variantes de faux-sens étaient les plus courantes (55 %) des variantes de codage ; Données étendues La figure 4 résume tous les effets de variantes prédits. Des variants délétères prédits par le score SIFT (Sorting Intolerant From Tolerant) ont été identifiés dans R3HCC1L , SH2B3 , CCDC171 , C1orf87 , LOXL4 , DLAT , ALG9 et SORT1. Dans l’ensemble de variantes crédibles, aucun gène n’était particulièrement intolérant à la variation fonctionnelle (le plus petit centile LoFtool (perte de fonction) était de 0,39). Pour les 42 locus associés, les cibles génétiques les plus probables de chacun ont été estimées par le score global V2G (variant-to-gene) d’OpenTargets (tableau supplémentaire 9 ). Deux variantes d’index (variante faux-sens rs12737449 ( C1orf87 ) et rs3735260 ( AUTS2 )) pourraient être causales car elles avaient des scores combinés d’épuisement dépendant de l’annotation (CADD) suggérant un effet délétère sur la fonction des gènes selon Kircher et al. 15 (tableau supplémentaire 10 ). L’ AUTS2la variante RegulomeDB de rang 2b indiquait un rôle régulateur ; son emplacement pris en charge par l’état de la chromatine sur un site de début de transcription actif 16 , 17 .
Sur les 173 gènes significatifs issus de tests basés sur les gènes à l’échelle du génome dans MAGMA (voir le tableau supplémentaire 11 pour leurs fonctions), 129 pourraient être annotés de manière fonctionnelle (tableau supplémentaire 12 ). Les séquences codant et non codant pour les protéines sont activement conservées dans environ les trois quarts de ces gènes, 63 % sont plus intolérants aux variations que la moyenne et 33 % sont intolérants aux mutations avec perte de fonction. L’analyse des propriétés génétiques des tissus généraux et de 13 tissus cérébraux a confirmé l’importance du cerveau et de régions cérébrales spécifiques (tableaux supplémentaires 13 et 14 ). Les niveaux d’expression cérébrale de 125 des 173 gènes significatifs issus de tests basés sur les gènes pourraient être cartographiés dans FUMA et sont présentés dans le tableau supplémentaire 15. Un total de 20 gènes ont montré des niveaux d’expression cérébrale généraux élevés et, parmi ceux-ci, 3 ( PPP1R1B, NPM1 et WASF3 ) étaient situés à proximité d’associations SNP significatives. Sur les 12 régions cérébrales évaluées, l’expression génique était généralement la plus élevée dans l’hémisphère cérébelleux, le cervelet et le cortex cérébral, conformément aux résultats de l’analyse des propriétés génétiques.
Héritabilité partitionnée
L’héritabilité de la dyslexie basée sur les SNP partitionnée par l’annotation fonctionnelle a montré un enrichissement significatif pour les régions conservées et les clusters H3K4me1 (tableau supplémentaire 16 et données étendues Fig. 5 ). Il y avait un enrichissement en gènes exprimés dans le cortex frontal, le cortex et le cortex cingulaire antérieur ( P <4,17 × 10 -3 ) (tableau supplémentaire 17 et données étendues Fig. 6 ), mais pas pour le type de cellule cérébrale (tableau supplémentaire 18 et données étendues Figure 7). Un enrichissement a été observé dans les régions activatrices et promotrices, identifiées par la présence de marques de chromatine H3K4me1 et H3K4me3, respectivement, dans plusieurs tissus du système nerveux central (SNC) (tableaux supplémentaires 19 et 20 et données étendues, figures 8 et 9 ). La lecture, une ramification du langage parlé, est un trait uniquement humain, mais il n’y a pas eu d’enrichissement pour une gamme d’annotations liées à l’évolution humaine couvrant les 30 derniers millions à 50 000 ans 18 (tableau supplémentaire 21 ).
Corrélations génétiques et LDSC
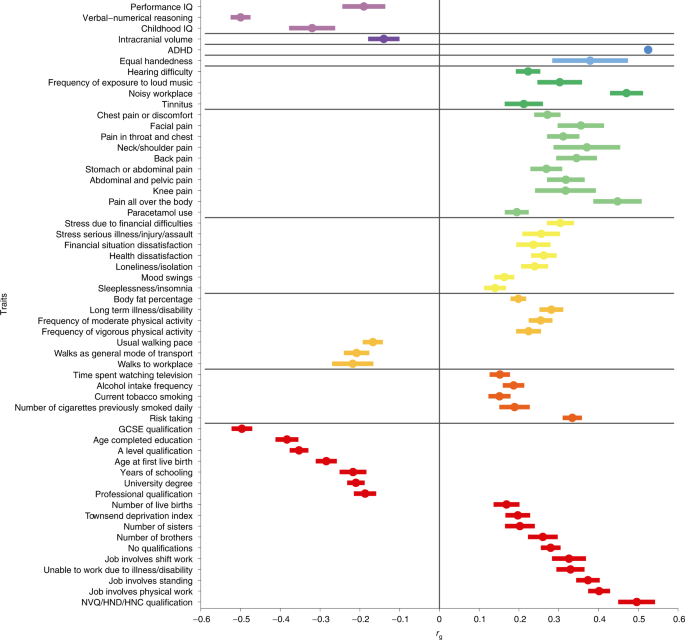
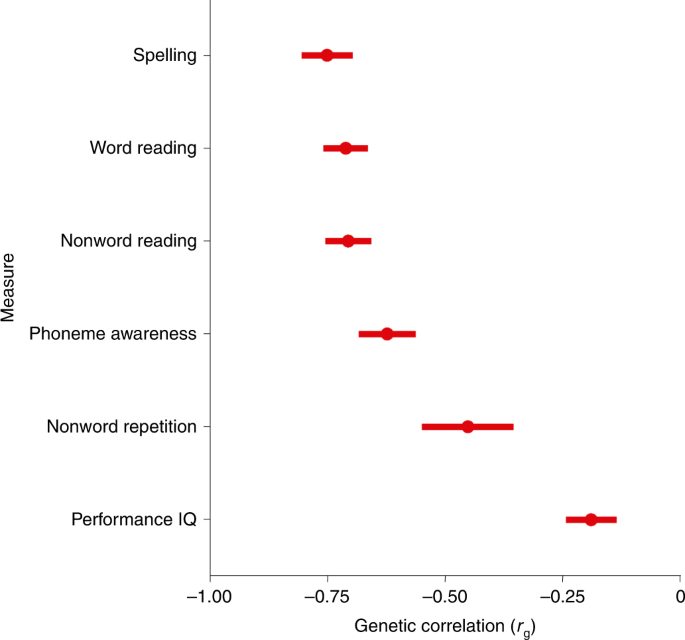
Les corrélations génétiques ont été estimées pour 98 traits (Fig. 2 et Tableau supplémentaire 22 ), y compris les mesures de lecture et d’orthographe, de GenLang (Fig. 3 ), et les volumes de structure sous-corticale cérébrale, la surface corticale totale et l’épaisseur de Enhancing Neuro Imaging Genetics via Consortium de méta-analyse (ENIGMA). Un total de 63 traits ont montré des corrélations génétiques avec la dyslexie au seuil de signification corrigé de Bonferroni ( P <0,05/98 ; Fig. 2 ). Corrélations génétiques ( r g) avec des mesures quantitatives de lecture et d’orthographe allant de -0,70 à -0,75 (IC à 95 % le plus bas de -0,60, IC à 95 % le plus élevé de -0,86) et étaient de -0,62 (IC à 95 % : -0,50, -0,74) et -0,45 (IC à 95 % : -0,26, -0,64) avec la conscience des phonèmes et les mesures de répétition des non-mots, respectivement. Le quotient intellectuel (QI) de performance (non verbal) chez l’enfant/adolescent était inférieur (-0,19 ; IC à 95 % : -0,08 , -0,30 ) à celui du raisonnement verbal-numérique chez l’adulte 19 (-0,50 ; IC à 95 % : -0,45 , -0,55) mais similaire à celle du QI de l’enfance 20 (-0,32 ; IC à 95 % : -0,21, -0,43) et du niveau d’instruction 21 (-0,22 ; IC à 95 % : -0,15, -0,29). Les traits montrant un r g positif incluaient les emplois impliquant un travail manuel lourd 21(0,40 ; (IC à 95 % : 0,34, 0,45)), qualifications liées au travail/professionnelles 21 (0,50 ; IC à 95 % : 0,41, 0,59), TDAH 22 (0,53 ; IC à 95 % : 0,29, 0,77), utilisation égale de mains droite et gauche 21 (0,38 ; IC 95 % : 0,19, 0,57) et mesure de la douleur 21 (moyenne = 0,31 ; IC 95 % : 0,21, 0,41). Sur les 11 mesures ENIGMA testées, seul le volume intracrânien était significativement corrélé à la dyslexie ( r g = −0,14 ; IC à 95 % : -0,06, -0,22). Une enquête ciblée sur 80 mesures de neuroimagerie structurelle de UK Biobank, y compris la morphométrie basée sur la surface et l’imagerie pondérée en diffusion pour les circuits cérébraux liés au langage, n’était pas significative à un niveau de signification corrigé de Bonferroni pour le nombre de traits indépendants. L’indépendance du phénotype a été estimée par décomposition spectrale de la matrice de corrélation phénotypique impliquée par l’interception LDSC bivariée à partir des statistiques récapitulatives GWAS de ces traits, à l’aide de la boîte à outils PhenoSpD 23 (tableau supplémentaire 23 ).
Analyses de scores polygéniques
Les scores polygéniques de dyslexie (PGS) basés sur le GWAS de dyslexie 23andMe ont été calculés dans quatre cohortes indépendantes et, dans l’ensemble, des PGS plus élevés étaient associés à une précision de lecture et d’orthographe plus faible (tableau supplémentaire 24 ). Dans deux échantillons basés sur la population australienne (1 647 adolescents, 1 163 adultes), le PGS de la dyslexie a expliqué jusqu’à 3,6 % de variance dans les mesures de lecture et d’orthographe, étant le plus prédictif d’une performance inférieure aux tests de lecture de non-mots, un indice de décodage phonologique. La dyslexie PGS n’était pas corrélée avec les scores aux tests de répétition de non-mots (considérés comme un marqueur de la mémoire phonologique à court terme). Dans les cohortes développementales enrichies pour les difficultés de lecture, le PGS dyslexie expliquait 3,7 % (UKdys ; n = 930) et 5,6 % (CLDRC ; n = 717) de variance dans les tests de reconnaissance de mots.
Analyses des associations dyslexiques à partir de la littérature
Sur 75 associations de dyslexie rapportées précédemment, aucune n’a montré de signification à l’échelle du génome dans nos analyses (tableau supplémentaire 25 ). Parmi ces variantes ciblées, 19 (dans ATP2C2 , CMIP , CNTNAP2 , DCDC2 , DIP2A , DYX1C1 , FOXP2 , KIAA0319L et PCNT ) ont montré une association survivant à la correction de Bonferroni qui expliquait la LD ( P < 0,05/68,7). Dans les tests basés sur les gènes de 14 gènes candidats de la littérature 24 , 25 , une association à un niveau de Bonferroni ( P < 0,05/14) a été observée pour KIAA0319L( P = 1,84 × 10 −4 ) et ROBO1 ( P = 1,53 × 10 −3 ) (Tableau supplémentaire 26 ). L’ association CNTNAP2 s’est approchée de la signification corrigée du niveau de réplication ( P = 0, 004). L’analyse ciblée de l’ensemble de gènes de trois voies précédemment impliquées dans la dyslexie (tableau supplémentaire 27 ) a montré un soutien au niveau de la réplication ( P = 2, 00 × 10 -3 ) pour la voie de guidage axonal (comprenant 216 gènes).
Discussion
Dans le plus grand GWAS de dyslexie à ce jour (> 50 000 diagnostics autodéclarés), nous avons identifié 42 locus indépendants significatifs. Parmi ceux-ci, 27 représentent de nouvelles associations qui n’ont pas été découvertes dans GWAS de traits cognitifs connexes ; 12 des nouvelles associations ont été validées dans la méta-analyse GWAS du consortium GenLang de la lecture/orthographe en anglais et dans d’autres langues européennes 14 , et 1 dans une cohorte de langue chinoise. Parmi les SNP significatifs, 36 % chevauchaient des variantes de la capacité cognitive générale GWAS, ce qui est cohérent avec des études jumelles qui montrent que la variation génétique du trouble de la lecture s’explique par la capacité cognitive générale et spécifique à la lecture 10. Semblable à d’autres traits complexes et compatible avec une polygénicité élevée, chaque locus significatif a montré de petits effets (rapports de cotes (OR) allant de 1,04 à 1,12). Notre héritabilité estimée basée sur le SNP de 19 % (en supposant une prévalence de 10 % de dyslexie dans la population) était égale à celle rapportée dans un GWAS plus petit 12 , mais inférieure aux estimations d’héritabilité des études sur les jumeaux (40 à 80 %) 26 , 27 . Cette différence peut être due en partie aux effets de variantes rares et structurelles 28 , qui ont été impliquées dans la lecture et les traits apparentés 29 , 30 .
Alors que AUTS2 a été impliqué dans l’autisme 31 , la déficience intellectuelle 32 et la dyslexie 33 , la variante que nous avons découverte (rs3735260) représente la plus forte association de SNP AUTS2 avec un trait neurodéveloppemental à ce jour. Parmi nos découvertes figuraient d’autres gènes connus du développement neurologique, tels que TANC2 (impliqué dans le retard de langage et la déficience intellectuelle 34 , 35 ) et, en particulier, GGNBP2 (lié au retard de développement neurologique 36 et à l’autisme 37) avec la variante rs34349354 prise en charge dans toutes nos cohortes de validation. Cependant, rs34349354 est également associé à la performance cognitive 38 et, sur la base des locus de traits quantitatifs d’expression (eQTL), la preuve est plus probablement liée à ZNHIT3 , colocalisant avec des QTL moléculaires ( opentargets.org ). Notamment, aucun des gènes candidats les plus établis pour la dyslexie n’a approché la signification à l’échelle du génome dans nos résultats.
Comme d’autres traits complexes humains, la partition de l’héritabilité basée sur les SNP a révélé un enrichissement dans les régions conservées 39 . Nous avons en outre observé un enrichissement dans la marque d’histone H3K4me1 (qui a également été signalée pour l’ASD 40 ), et dans les grappes H3K4me1 et H3K4me3 dans le SNC (marquage des activateurs et des promoteurs, respectivement). Étant donné que les systèmes de lecture/écriture sont construits sur nos capacités de langage parlé, il est plausible que les changements évolutifs de la lignée humaine aient contribué à façonner l’architecture génétique sous-jacente 41 . Cependant, nous n’avons pas trouvé d’enrichissement d’associations significatives pour les annotations organisées couvrant différentes périodes de la préhistoire des hominines.
Notre trait binaire de diagnostic de dyslexie autodéclaré a montré de fortes corrélations génétiques négatives avec des mesures quantitatives de lecture et d’orthographe, soutenant la validité de cette mesure dans la cohorte 23andMe et suggérant que les compétences en lecture et les troubles ne sont pas qualitativement distincts. La corrélation génétique positive entre les difficultés auditives et la dyslexie est cohérente avec les corrélations génétiques rapportées pour les compétences en lecture pendant l’enfance 42 , suggérant que les problèmes auditifs à un âge précoce pourraient affecter l’acquisition des compétences de traitement phonologique.
La dyslexie a montré des corrélations génétiques modérément négatives avec le raisonnement verbal-numérique adulte, mais il y avait un manque de corrélation génétique forte de la dyslexie avec le QI de performance (non verbal). Cela serait cohérent avec les observations phénotypiques selon lesquelles les personnes atteintes de dyslexie sont désavantagées lors des tests de QI verbaux 43 . Les corrélations du niveau de scolarité n’étaient pas non plus fortes, ce qui pourrait refléter des ajustements scolaires et d’autres soutiens qui contrecarrent le désavantage dans l’apprentissage scolaire.
Il y avait peu de preuves que la variation génétique commune de la dyslexie soit liée aux différences interindividuelles dans les volumes sous-corticaux, ou à la connectivité structurelle et à la morphométrie des régions cérébrales impliquées dans le traitement du langage chez les adultes. Ainsi, les corrélations phénotypiques précédemment rapportées entre la dyslexie et certains aspects de la neuroanatomie peuvent en grande partie refléter la formation environnementale du cerveau, peut-être à travers le processus de lecture lui-même 44 . La gaucherie et l’ambidextrie présentent un léger chevauchement génétique 45 , mais sont toutes deux phénotypiquement liées à des troubles neurodéveloppementaux/capacités cognitives 46 , 47. Nous rapportons une corrélation génétique significative entre la dyslexie et l’utilisation égale des mains auto-déclarée, mais pas la gaucherie, soutenant les théories liant l’ambidextrie et la dyslexie 48 .
La dyslexie et le TDAH 5 , 6 coexistent souvent (24 % déclarant un TDAH dans nos cas contre 9 % chez les témoins), et nous montrons une corrélation génétique modérée entre les deux, reflétant potentiellement des endophénotypes partagés comme des déficits de la mémoire de travail et de l’attention 49 . Bien que nous n’ayons pas trouvé de corrélations génétiques significatives entre la dyslexie et le TSA, le GWAS pour ce dernier englobait divers phénotypes neurodéveloppementaux, y compris des sous-groupes avec un niveau d’instruction et un QI variables 40 . Les corrélations génétiques avec les traits liés à la douleur suggèrent que les personnes dyslexiques peuvent avoir un seuil inférieur de perception de la douleur. Des liens entre la douleur et d’autres troubles neurodéveloppementaux ont été rapportés 50 , 51 .
Les scores polygéniques de dyslexie étaient corrélés avec des résultats inférieurs aux tests de lecture et d’orthographe dans des échantillons basés sur la population et enrichis en troubles de lecture, en particulier pour la lecture de non-mots, une mesure du décodage phonologique qui est généralement altérée dans la dyslexie. Les scores polygéniques pourraient devenir un outil précieux pour aider à identifier les enfants ayant une propension à la dyslexie, permettant un soutien à l’apprentissage avant le développement des compétences en lecture. Cependant, une limite de notre étude est le potentiel de biais de collision résultant de la sélection de l’échantillon (c’est-à-dire des personnes sans dyslexie et de positions socio-économiques plus élevées), que nous n’avons pas été en mesure de quantifier ; ainsi, des précautions doivent être prises dans les recherches futures lors de l’utilisation de scores polygéniques basés sur de nombreuses variantes 52 .
En résumé, nous rapportons 42 nouveaux locus significatifs indépendants à l’échelle du génome associés à la dyslexie, dont 27 n’ont pas été associés à des traits cognitivo-éducatifs et devraient être prioritaires pour le suivi en tant que candidats à la dyslexie. L’annotation fonctionnelle des variants met en évidence l’importance des régions conservées et amplificatrices du génome pour ce trait. La dyslexie montre des corrélations génétiques positives avec le TDAH, les qualifications professionnelles, les occupations physiques, l’ambidextrie et la perception de la douleur, et des corrélations négatives avec les qualifications académiques et les capacités cognitives ; des méthodes basées sur la famille sont nécessaires pour dissocier les effets pléiotropiques et causaux.
Méthodes
Participants GWAS
Les participants provenaient de la clientèle de 23andMe, Inc., une société de génétique grand public. Les participants ont fourni un consentement éclairé et ont participé à la recherche en ligne, selon un protocole approuvé par l’IRB externe accrédité par l’AAHRPP, Ethical and Independent Review Services ( www.eandireview.com). Ils comprenaient 51 800 participants (21 513 hommes, 30 287 femmes) qui ont répondu « oui » à la question « Avez-vous reçu un diagnostic de dyslexie ? » (cas) et 1 087 070 (446 054 hommes, 641 016 femmes) participants qui ont répondu « non » (témoins). L’âge variait de 18 à 110 ans, la prévalence de la dyslexie étant plus élevée chez les jeunes participants (5,34 % chez les 20 à 30 ans) que chez les participants plus âgés (3,23 % chez les 80 à 90 ans). La relation linéaire négative entre la prévalence de la dyslexie et l’âge des participants était attendue étant donné que le dépistage de difficultés d’apprentissage spécifiques n’est devenu courant qu’au cours des dernières décennies. De plus, cela concorde avec les résultats du sous-échantillon (4,3 %) de participants qui ont déclaré l’âge du diagnostic : les participants plus jeunes ont été diagnostiqués à un âge plus précoce (par exemple, 9,7 ans (±4. 7) pour les 20 à 30 ans) que les participants plus âgés (par exemple, 22,4 ans (±17,8) pour les 80 à 90 ans). La prévalence de la dyslexie dans notre échantillon était similaire chez les femmes (4,51 %) et les hommes (4,6 %), bien que la prévalence légèrement plus élevée chez les hommes dans ce très grand échantillon soit statistiquement significative (P < 8,7 × 10 -6 ). Une telle prévalence se situe à l’extrémité inférieure de la fourchette généralement signalée dans la population américaine 3 et pourrait représenter les cas les plus graves de dyslexie étant donné qu’un diagnostic formel était requis ; De plus, les personnes atteintes de dyslexie pourraient se retirer des sondages qui nécessitent de la lecture, ce qui restreindrait davantage l’éventail de l’échantillon.
Génotypage et imputation
L’ADN a été extrait d’échantillons de salive et génotypé sur l’une des cinq plates-formes de génotypage de l’Institut national de génétique (NGI). Dans la présente analyse, seuls les participants d’ascendance européenne ont été inclus. Des détails sur les matrices de génotypage, le contrôle de la qualité des échantillons et la dérivation de l’ascendance peuvent être trouvés dans Fontanillas et al. 53 et la note complémentaire . Les génotypes en phase ont été imputés à un panel de référence combiné des haplotypes de phase 3 de 1000 génomes (mai 2015) et au panel de référence d’imputation UK10K à l’aide de Minimac3 (voir Das et al. 54 ).
Analyse des associations
Une analyse d’association a été effectuée sur des données de dosage de SNP génotypées et imputées à l’aide d’une régression logistique et en supposant un modèle additif d’effets alléliques. Pour l’analyse du chromosome X, les génotypes mâles ont été traités comme des diploïdes homozygotes. Les covariables comprenaient l’âge, l’âge au carré, le sexe, les cinq premières composantes principales de l’ascendance et la plate-forme génotypique. La signification du SNP a été évaluée par un test de rapport de vraisemblance et la signification à l’échelle du génome a été déterminée comme étant P < 5 × 10 −8 (niveau de signification suggestif comme P < 1 × 10 −6 ). Seuls les SNP imputés de manière fiable ( r 2 > 0,80) et ceux dont la fréquence des allèles mineurs (MAF) > 0,01 sont présentés ( n = 7 995 923). Nous définissons les régions associées en identifiant d’abord tous les variants avec P < 5 × 10 −8 , puis en regroupant ces variants en régions séparées par des espaces d’au moins 250 kb. Les variantes d’index sont les variantes avec la plus petite valeur P dans chaque région associée. Nous utilisons la même approche pour les régions avec des associations suggestives, mais en identifiant d’abord tous les variants avec P < 10 −5 . Analyse subsidiaire d’association à l’échelle du génome de groupes séparés d’hommes ( n = 21 513 cas, 446 054 témoins) et de femmes ( n = 30 287 cas, 641 016 témoins), et plus jeunes (moins de 55 ans; n = 30 763 cas, 582 276 témoins) et plus âgés (55 et ci-dessus;n = 21 037 cas, 504 794 groupes témoins) a été réalisée. Ce dernier consistait à vérifier si la fiabilité du diagnostic (supposée plus élevée dans l’échantillon plus jeune dont le rappel du diagnostic devrait être meilleur et qui aurait été exposé à des niveaux plus élevés de dépistage de la dyslexie) affectait le signal GWAS.
Nous avons également cherché à valider indépendamment nos variantes significatives à l’échelle du génome dans (1) une méta-analyse GWAS publiée de 2 274 cas de dyslexie de neuf pays européens représentant six langues différentes (NeuroDys) par Gialluisi et al. 55 ; (2) un échantillon de population (étude chinoise sur la lecture ; CRS) d’enfants mesurés sur des traits quantitatifs de précision et de fluidité de lecture ( n = 2 270 ; décrit dans la note supplémentaire ) et ; (3) dans la méta-analyse GWAS des traits quantitatifs GenLang de la lecture de mots (jusqu’à n = 33 959) et de l’orthographe (jusqu’à n = 18 514) compétences mesurées dans des cohortes d’enfants et d’adolescents d’Europe, des États-Unis et d’Australie, et représentant sept langues européennes, dont l’anglais était la plus répandue 14 .
Analyses basées sur les gènes
Les résultats GWAS ont été utilisés pour calculer les valeurs P basées sur les gènes pour l’association avec la dyslexie en effectuant l’analyse génétique dans MAGMA v.1.08 (réf. 56 ) via l’interface FUMA 57 en utilisant des paramètres standard. Au total, 19 039 gènes ont été testés et les valeurs P ont été jugées sur la base d’un seuil de signification corrigé de Bonferroni de P < 2,63 × 10 −6 . Nous avons également effectué des analyses d’ensembles de gènes pour l’association de voies biologiques (tous les termes d’ontologie génique (GO) disponibles et les ensembles de gènes sélectionnés à partir de la base de données de signatures moléculaires (MsigDB) 58 , 59 ) avec la dyslexie dans MAGMA via l’interface FUMA. Le nombre total de voies testées était de 15 486, etLes valeurs P ont été jugées sur la base d’un seuil de signification corrigé de Bonferroni de P < 3,23 × 10 −6 .
Annotations biologiques
Les variants significatifs à l’échelle du génome et les gènes voisins ont été annotés à l’aide de données de référence externes et évalués pour leur impact fonctionnel ou réglementaire. Un ensemble crédible à 99 % de variantes potentiellement causales pour les SNP dans des régions significatives était basé sur le facteur Bayes approximatif (ABF) 60 en supposant une variance antérieure de 0,1 et en utilisant la méthode de Maller et al. 61 pour définir ces ensembles. La prédiction de l’effet de variante de ceux-ci a été réalisée dans ENSEMBL (version 104) 62. Pour les variants significatifs à l’échelle du génome, nous avons pris en compte : le contexte génique (si un variant est intergénique ou situé dans une région fonctionnelle spécifique au sein d’un locus génique) ; la délétère (score Combiné Annotation Dependent Depletion (CADD)); fonctionnalité (catégorie RegulomeDB (RDB)); état de chromatine (état de chromatine minimum et commun à 15 noyaux); et les associations SNP-trait signalées dans le catalogue NHGRI GWAS 13 .
Pour chaque variante, la cible génétique la plus probable a été identifiée à l’aide du portail Open Target Genetics 63, qui s’appuie sur des preuves issues d’expériences d’interaction QTL et chromatine, de prédictions fonctionnelles et de la distance par rapport au site de début de transcription d’un gène. Pour les gènes significatifs à l’échelle du génome, nous avons pris en compte : l’intolérance à la perte de fonction (probabilité de score d’intolérance à la perte de fonction (pLI) ); intolérance à la variation (score résiduel d’intolérance à la variation, RVIS); intolérance à la variation dans les régions non codantes (RVIS non codant, ncRVIS); contrainte évolutive des régions non codantes (score noncoding genomic evolutionary rate profiling (ncGERP)) ; contrainte évolutive des régions codant pour les protéines (score de profilage du taux d’évolution génomique codant pour les protéines (pcGERP)); effet délétère dans les régions non codantes (score CADD non codant (ncCADD) ); fonctionnalité combinée des variants dans les régions non codantes (score d’annotation des variants à l’échelle du génome non codant (ncGWAVA) ); et expression dans 12 tissus cérébraux (amygdale, cortex cingulaire antérieur, noyaux gris centraux caudés, hémisphère cérébelleux, cervelet, cortex, cortex frontal, hippocampe, hypothalamus, noyau accumbens noyaux gris centraux, ganglions basaux putamen et substantia nigra). Toutes les annotations ont été obtenues via FUMA57 sauf RVIS, ncGERP, pcGERP, ncCADD et ncGWAVA, qui sont tirés de Petrovski et al. 64 . Les détails de chaque annotation, y compris les sources originales, se trouvent dans la note supplémentaire .
Héritabilité partitionnée
Nous avons partitionné l’héritabilité SNP de la dyslexie en utilisant le LDSC stratifié, comme décrit par Finucane et al. 39 , pour déterminer si les SNP qui partagent la plus grande proportion de l’héritabilité sont également regroupés dans des catégories fonctionnelles spécifiques du génome. Dans l’ensemble, nous avons effectué 266 tests différents, ce qui donnerait un niveau de signification corrigé de Bonferroni très conservateur de 1,88 × 10 −4 , mais comme il y aura un chevauchement entre les groupes d’annotations, nous rapportons également des corrections de signification dans différentes classes d’annotations, chacune de que nous décrivons maintenant. Le partitionnement a été effectué pour les 24 principales annotations fonctionnelles définies par Finucane et al. 39 . Les scores LD, les poids de régression et les fréquences alléliques proviennent d’échantillons d’ascendance européenne et ont été extraits dehttps://alkesgroup.broadinstitute.org/LDSCORE . Les estimations de l’héritabilité étaient considérées comme statistiquement significatives si la valeur P dépassait un niveau α de 2,08 × 10 −3 , dérivé par la correction de Bonferroni basée sur 24 tests.
Nous avons également estimé l’enrichissement de l’héritabilité de la dyslexie pour les annotations spécifiques aux tissus, tout en contrôlant les annotations dans le modèle de base, y compris l’expression génique dans trois types de cellules cérébrales, l’expression génique dans 12 régions cérébrales et les marques de chromatine H3K4me1 et H3K4me3 dans plusieurs tissus. (108 et 114, respectivement) puisque ces marques sont enrichies au niveau des activateurs 65 et des promoteurs 66 , respectivement. L’enrichissement est la proportion d’héritabilité des SNP divisée par la proportion de SNP. Pour les types de cellules cérébrales, nous avons estimé l’enrichissement pour l’héritabilité de la dyslexie pour les gènes exprimés dans les neurones, les astrocytes et les oligodendrocytes en utilisant les données de Cahoy et al. 67 . Les enrichissements ont été considérés comme statistiquement significatifs si le Pla valeur a dépassé un niveau α de 0,017, dérivé par la correction de Bonferroni basée sur trois tests. Les données d’expression génique utilisées pour estimer l’enrichissement de l’héritabilité dans les gènes exprimés dans certaines régions du cerveau provenaient de la base de données GTEx 68 , et le niveau α dérivé de Bonferroni pour l’enrichissement était de 4,17 × 10 -3 (basé sur 12 tests). Les annotations de chromatine incluent des données du consortium Roadmap Epigenomics 17 et EN-TEX 69 , 70 . Pour H3K4me1, le niveau α dérivé de Bonferroni pour l’enrichissement était de 4,63 × 10 −4 (sur la base de 108 tests) et, pour H3K4me3, le niveau α dérivé de Bonferroni pour l’enrichissement était de 4,39 × 10 −4 (sur la base de 114 tests).
Annotations évolutives
Bien que la lecture et l’écriture soient une invention culturelle humaine, elles s’appuient sur des voies fondamentales impliquées dans le traitement du langage. Par conséquent, nous avons cherché à savoir si les annotations liées à l’évolution humaine étaient significativement enrichies pour l’héritabilité de la dyslexie en appliquant un pipeline d’analyse évolutive adapté de Tilot et al. 18 . Ces analyses capturent une gamme de périodes dans un calendrier évolutif sur la lignée qui a conduit à l’homme, d’il y a environ 30 millions d’années à 50 000 ans.
L’enrichissement de l’héritabilité a été estimé dans les amplificateurs acquis par l’homme du cerveau adulte (HGE) 71 , les HGE du cerveau fœtal 72 , les anciennes régions de balayage sélectif 73 , les SNP introgressés par les Néandertaliens 74 et les régions appauvries par les Néandertaliens 75 (voir la note supplémentaire pour une description de chaque annotation) ; et contrôlé pour l’utilisation du modèle de base LD v.2 de Gazal et al. 76 . L’enrichissement de l’héritabilité dans les HGE humains adultes et fœtaux a également été contrôlé pour les éléments régulateurs actifs du cerveau adulte et fœtal à partir de la ressource Roadmap Epigenomics 17 . Les éléments régulateurs actifs ont été définis à l’aide de chromHMM 16 . Enrichissement Ples valeurs ont été jugées par un niveau α de 10 -2 , dérivé par la correction de Bonferroni basée sur cinq tests.
Corrélations génétiques
Corrélations génétiques au sein du 23andMe GWAS de la dyslexie
La corrélation génétique entre le diagnostic de dyslexie autodéclarée chez les hommes et les femmes, et entre les adultes plus jeunes (<55 ans) et plus âgés (≥55 ans) a été calculée à l’aide du LDSC 77 , 78 .
Corrélations génétiques de la dyslexie avec d’autres traits
Nous présentons la corrélation génétique par paires de la dyslexie avec 98 traits. Les statistiques récapitulatives pour la plupart de ces traits sont accessibles au public via LD Hub 77 , 78 , 79 – une base de données centralisée et une interface Web qui automatise le pipeline d’analyse de régression LDSC. Une sélection de mesures d’imagerie par résonance magnétique cérébrale obtenues auprès du consortium ENIGMA-3 80 , 81 , 82 , 83 , et des mesures de la précision de la lecture et de l’orthographe et du QI de performance du GenLang Consortium 14ont été analysés localement à l’aide de LDSC. La précision de la lecture des mots dans GenLang a été mesurée par le nombre de mots corrects lus à haute voix à partir d’une liste de manière limitée ou non. Des exemples d’outils qui incluent cette mesure sont le Test of Word Reading Efficiency (TOWRE), les British Ability Scales (BAS) et le Wide Range Achievement Test (WRAT). La précision orthographique dans GenLang a été mesurée par le nombre de mots correctement orthographiés oralement ou par écrit. Les mots ont été dictés sous forme de mots simples ou dans une phrase. Des exemples d’outils qui incluent cette mesure sont les dimensions BAS, WRAT et Wechsler Objective Reading Dimensions (WORD). Le QI de performance dans GenLang était basé sur des sous-tests de tests de QI qui ne dépendaient pas d’indices verbaux, tels qu’inclus par exemple dans le BAS et l’échelle d’intelligence de Wechsler pour les enfants (WISC). 22 . La correction de Bonferroni pour les tests multiples a dérivé une valeur P critique ajustée de 5,1 × 10 −4 à partir de 98 tests indépendants.
Les corrélations génétiques ont été estimées plus en détail dans une analyse ciblée des mesures d’imagerie par résonance magnétique cérébrale structurelle de UK Biobank, qui étaient plus complètes que celles actuellement disponibles auprès d’ENIGMA, ainsi que d’autres avantages tels que des données spécifiques à l’hémisphère et une plus grande homogénéité dans les procédures de cohorte et de numérisation. Les statistiques sommaires du GWAS à partir des phénotypes dérivés de l’imagerie cérébrale pour 33 000 participants ont été téléchargées à partir de l’Oxford Brain Imaging Genetics Server 84 . Les traits d’imagerie cérébrale structurelle englobaient à la fois l’imagerie du tenseur de diffusion et les phénotypes morphométriques basés sur la surface 85où les secteurs ou les régions d’intérêt sélectionnés avaient un lien connu avec la langue. Pour l’imagerie du tenseur de diffusion, les valeurs d’anisotropie fractionnelle dérivées à la fois des statistiques spatiales basées sur les voies et de la tractographie probabiliste ont été utilisées pour les voies disponibles couvrant le réseau linguistique étendu 86 . Pour le GWAS morphométrique basé sur la surface (volume cortical, surface et épaisseur), des statistiques récapitulatives pour les régions d’intérêt dérivées de l’atlas Desikan-Killiany (surface blanche) ont été utilisées, à nouveau sélectionnées pour leur pertinence dans le traitement du langage, sur la base de la littérature précédente 87 , 88 , 89 , 90 . Pour corriger les tests multiples, les corrélations phénotypiques entre les indices d’imagerie de la UK Biobank ont été dérivées et analysées par PhenoSpD23 pour obtenir le nombre de variables indépendantes (36,08) à utiliser pour la correction de Bonferroni ( valeur P critique ajustée de 1,39 × 10 −3 ).
Analyses de scores polygéniques
Les scores de dyslexie polygénique étaient basés sur un nombre de plus en plus grand de SNP correspondant à leurs valeurs d’ association P du 23andMe GWAS ( P < 5 × 10 −8 , P < 1 × 10 −5 , P < 0,001, P < 0,01, P < 0,05, P < 0,1, P < 0,5, 1). Ils ont été calculés dans quatre cohortes indépendantes. Deux étaient des cohortes de la population générale d’Australie : n = 1 640 (772 familles) adolescents/jeunes adultes (adolescents de Brisbane) 91 ; n = 1 165 (966 familles) adultes plus âgés (adultes de Brisbane) 25. Les deux autres étaient des échantillons familiaux sélectionnés pour la dyslexie : un du Royaume-Uni (UKdys), n = 930 (595 familles) ; l’autre des États-Unis (Colorado Learning Disabilities Research Center, CLDRC), n = 717 (336 familles) 92 . Dans les échantillons australiens, les scores polygéniques ont été calculés sur 1 000 données génétiques imputées de phase 3 de génomes (v.20101123) à l’aide de PLINK 93 . Seuls les SNP imputés de manière fiable ( R 2 > 0,80) et ceux avec une fréquence d’allèles mineurs > 0,01 ont été inclus, et la procédure d’agrégation par défaut a été utilisée lorsque les SNP index formaient un amas avec d’autres SNP dans LD ( R 2 > 0,1) et dans un rayon de 250 kb. Dans les échantillons UKdys et CLDRC, les scores polygéniques ont été calculés sur les données génétiques imputées du Haplotype Reference Consortium à l’aide de PRSice 94 , avec la même qualité d’imputation et les mêmes exclusions MAF pour l’échantillon de base (23andMe GWAS) et les paramètres d’agrégation.
Les scores polygéniques ont ensuite été utilisés comme prédicteurs dans des modèles linéaires de résultats de traits quantitatifs (Australie : tests de lecture et d’orthographe de mots, de non-mots (phonétiques), de mots irréguliers (lexicaux) à partir d’une version étendue du Components of Reading Examination 95 , et deux tests de répétition de non-mots qui sont sensibles aux troubles développementaux du langage—Dollaghan et Campbell 96 , Gathercole et Baddeley 97; UKdys et CLDRC : reconnaissance de mots). Tous les traits quantitatifs ont été préajustés pour les composantes principales du sexe, de l’âge et de l’ascendance (10 composantes principales dans UKdys et CLDR ; 20 composantes principales dans les échantillons australiens). D’autres ajustements ont été effectués pour le cycle d’imputation (séries séparées pour différents tableaux de génotypage) dans les échantillons australiens, et pour le QI non verbal dans tous les échantillons (à l’exception des adultes australiens) et pour les difficultés auditives chez les adultes âgés australiens. Étant donné que les cohortes comprenaient des membres de la famille apparentés (jumeaux ou frères et sœurs), des modèles linéaires mixtes (lme) ont été spécifiés dans RStudio 98 , l’appartenance à la famille étant modélisée comme un effet aléatoire et le score polygénique de dyslexie comme un effet fixe. Lorsque des jumeaux monozygotes étaient présents, leurs scores de traits ont été moyennés et ils ont été utilisés comme un seul cas.
Évaluation des candidats de la littérature précédente
Nous avons utilisé les résultats du 23andMe dyslexia GWAS pour évaluer les variantes, les gènes et les voies biologiques précédemment associés ou impliqués dans la dyslexie et/ou la variation de la capacité de lecture et d’orthographe dans des études d’association passées, des analyses de liaison et d’autres études.
Variantes précédemment signalées
Nous avons évalué 75 variantes précédemment rapportées dans nos statistiques récapitulatives, en adoptant un seuil de signification de réplication/validation de P < 7,28 × 10 −4 , dérivé par correction de Bonferroni basé sur 68,7 tests indépendants dérivés par décomposition spectrale matricielle, en tenant compte de LD (voir Doust et al. 25 pour plus de détails sur la manière dont ces variantes ont été sélectionnées). Les sources de chaque variante sont fournies dans le tableau supplémentaire 26 .
Gènes candidats de la dyslexie
Nous avons évalué les résultats basés sur les gènes de MAGMA v.1.08 (réf. 56 ) pour la surreprésentation des variantes significatives à l’échelle du génome du GWAS de la dyslexie 23andMe dans les loci de 14 gènes candidats de la littérature antérieure : CMIP , CNTNAP2 , CYP19A1 , DCDC2 , DIP2A , DYX1C1 , GCFC2 , KIAA0319 , KIAA0319L , MRPL19 , PCNT , PRMT2 , S100B et ROBO1 . La justification de cette sélection est détaillée par Luciano et al. 24 et Doust et al. 5. La valeur P critique , basée sur la correction de Bonferroni pour 14 tests, était de 3,57 × 10 −3 .
Ensembles de gènes candidats à la dyslexie
Nous avons effectué une analyse d’ensemble de gènes dans MAGMA pour tester la surreprésentation de variants significatifs à l’échelle du génome dans (1) un ensemble de cibles transcriptionnelles de FOXP2 , un facteur de transcription hautement conservé lié aux troubles de la parole et du langage 99 ; et (2) deux voies biologiques précédemment suggérées pour jouer un rôle dans la susceptibilité à la dyslexie 100 , 101 – le guidage axonal (GO:0007411 : ‘processus de chimiotaxie qui dirige la migration d’un cône de croissance axonale vers un site cible spécifique’ ; 216 gènes) et migration des neurones (GO:0001764 : « « » 145 gènes. Un P critique ajustéLa valeur de 0,017 a été dérivée en utilisant la correction de Bonferroni basée sur trois tests indépendants.
Normes éthiques
Les participants ont fourni un consentement éclairé et ont participé à la recherche en ligne, selon un protocole approuvé par l’IRB, les services d’examen éthique et indépendant accrédités par l’AAHRPP. Les participants ont été inclus dans l’analyse sur la base du statut de consentement tel qu’il a été vérifié au moment où les analyses de données ont été lancées.
Résumé des rapports
De plus amples informations sur la conception de la recherche sont disponibles dans le résumé des rapports de recherche sur la nature lié à cet article.
Disponibilité des données
Les statistiques récapitulatives complètes pour chaque dyslexie GWAS présentées dans cet article seront mises à disposition sur le site Web de 23andMe ( https://research.23andme.com/dataset-access/ ) aux chercheurs qualifiés dans le cadre d’un accord avec 23andMe qui protège la confidentialité de 23andMe. participants. Les 10 000 principaux SNP associés du principal GWAS peuvent être téléchargés à partir de https://doi.org/10.7488/ds/3465 .
Références
- Ritchie, SJ & Bates, TC Liens durables entre les mathématiques de l’enfance et la réussite en lecture au statut socio-économique adulte. Psychol. Sci. 24 , 1301-1308 (2013).Article PubMed Google Scholar
- Shaywitz, SE, Shaywitz, BA, Fletcher, JM & Escobar, MD Prévalence des troubles de la lecture chez les garçons et les filles : résultats de l’étude longitudinale du Connecticut. JAMA 264 , 998-1002 (1990).Article CAS PubMed Google Scholar
- Katusic , SK , Colligan , RC , Barbaresi , WJ , Schaid , DJ et Jacobsen , SJ Mayo Clin. Proc. 76 , 1081-1092 (2001).Article CAS PubMed Google Scholar
- Carroll, JM, Maughan, B., Goodman, R. & Meltzer, H. Difficultés d’alphabétisation et troubles psychiatriques : preuves de comorbidité. J. Child Psychol. Psychiatrie 46 , 524–532 (2005).Article PubMed Google Scholar
- Margari, L. et al. Comorbidités neuropsychopathologiques dans les troubles des apprentissages. BMC Neurol. 13 , 198 (2013).Article PubMed Centre PubMed Google Scholar
- Willcutt, EG, Pennington, BF & DeFries, JC Étude jumelle de l’étiologie de la comorbidité entre le trouble de lecture et le trouble déficitaire de l’attention/hyperactivité. Un m. J. Med. Genet. 96 , 293-301 (2000).Article CAS PubMed Google Scholar
- McArthur, GM, Hogben, JH, Edwards, VT, Heath, SM & Mengler, ED Sur les « spécificités » d’un trouble de lecture spécifique et d’un trouble du langage spécifique. J. Child Psychol. Psychiatrie 41 , 869–874 (2000).Article CAS PubMed Google Scholar
- Catts, HW, Fey, ME, Tomblin, JB et Zhang, X. Une enquête longitudinale sur les résultats en lecture chez les enfants ayant des troubles du langage. J. Discours Lang. Écouter. Rés. 45 , 1142–1157 (2002).Article PubMed Google Scholar
- Bates, TC et al. Bases génétiques et environnementales de la lecture et de l’orthographe : un modèle génétique unifié à double voie. Lis. Ordonnance. 20 , 147-171 (2007).Article Google Scholar
- Haworth, CMA et al. Gènes généralistes et troubles d’apprentissage : une analyse génétique multivariée des faibles performances en lecture, en mathématiques, en langage et en capacités cognitives générales dans un échantillon de 8 000 jumeaux de 12 ans. J. Child Psychol. Psychiatrie 50 , 1318–1325 (2009).Article PubMed Centre PubMed Google Scholar
- Fisher, SE & DeFries, JC Dyslexie développementale : dissection génétique d’un trait cognitif complexe. Nat. Rév. Neurosci. 3 , 767–780 (2002).Article CAS PubMed Google Scholar
- Gialluisi, A. et al. Une étude d’association à l’échelle du génome révèle de nouvelles informations sur l’héritabilité et les corrélats génétiques de la dyslexie développementale. Mol. Psychiatrie 26 , 3004–3017 (2021).Article CAS PubMed Google Scholar
- Buniello, A. et al. Le catalogue NHGRI-EBI GWAS des études d’association à l’échelle du génome publiées, des tableaux ciblés et des statistiques récapitulatives 2019. Nucleic Acids Res. 47 , D1005–D1012 (2018).Article Centre PubMed Google Scholar
- Eising, E. et al. Analyses à l’échelle du génome des différences individuelles dans les compétences liées à la lecture et au langage évaluées quantitativement chez jusqu’à 34 000 personnes. Proc. Natl Acad. Sci. États-Unis 119 , e2202764119 (2022).Article CAS PubMed Centre PubMed Google Scholar
- Kircher, M. et al. Un cadre général pour estimer la pathogénicité relative des variants génétiques humains. Nat. Genet. 46 , 310-315 (2014).Article CAS PubMed Centre PubMed Google Scholar
- Ernst, J. & Kellis, M. ChromHMM : automatisation de la découverte et de la caractérisation de l’état de la chromatine. Nat. Méthodes 9 , 215–216 (2012).Article CAS PubMed Centre PubMed Google Scholar
- Kundaje, A. et al. Analyse intégrative de 111 épigénomes humains de référence. Nature 518 , 317–330 (2015).Article CAS PubMed Centre PubMed Google Scholar
- Tilot, AK et al. L’histoire évolutive des variantes génétiques communes influençant la surface corticale humaine. Cortex cérébral 31 , 1873–1887 (2020).Article Centre PubMed Google Scholar
- Sniekers, S. et al. Une méta-analyse d’association à l’échelle du génome de 78 308 individus identifie de nouveaux locus et gènes influençant l’intelligence humaine. Nat. Genet. 49 , 1107-1112 (2017).Article CAS PubMed Centre PubMed Google Scholar
- Benyamin, B. et al. L’intelligence de l’enfant est héréditaire, hautement polygénique et associée à FNBP1L. Mol. Psychiatrie 19 , 253–258 (2014).Article CAS PubMed Google Scholar
- Bycroft, C. et al. La ressource UK Biobank avec un phénotypage approfondi et des données génomiques. Nature 562 , 203-209 (2018).Article CAS PubMed Centre PubMed Google Scholar
- Middeldorp, CM et al. Une méta-analyse d’association à l’échelle du génome des symptômes du trouble déficitaire de l’attention / hyperactivité dans des cohortes pédiatriques basées sur la population. Confiture. Acad. Enfant Adolescentc. Psychiatrie 55 , 896–905.e6 (2016).Article PubMed Centre PubMed Google Scholar
- Zheng, J. et al. PhenoSpD : une boîte à outils intégrée pour l’estimation de la corrélation phénotypique et la correction de tests multiples à l’aide de statistiques récapitulatives GWAS. Gigascience7 , giy090 (2018).Article Centre PubMed Google Scholar
- Luciano, M., Gow, AJ, Pattie, A., Bates, TC et Deary, IJ L’influence des gènes candidats de la dyslexie sur les compétences en lecture chez les personnes âgées. Comportement Genet. 48 , 351–360 (2018).Article PubMed Centre PubMed Google Scholar
- Doust, C. et al. L’association de la dyslexie et des gènes candidats des troubles développementaux de la parole et du langage avec les capacités de lecture et de langage chez les adultes. Rés jumelle. Hum. Genet. 23 , 23–32 (2020).Article PubMed Google Scholar
- Davis, CJ, Knopik, VS, Olson, RK, Wadsworth, SJ & DeFries, JC Génétique et influences environnementales sur la capacité de dénomination rapide et de lecture. Ann. Dyslexie 51 , 231–247 (2001).Article Google Scholar
- Gayán, J. & Olson, RK Influences génétiques et environnementales sur les compétences orthographiques et phonologiques chez les enfants ayant des troubles de lecture. Dév. Neuropsychol. 20 , 483–507 (2001).Article PubMed Google Scholar
- Hannula-Jouppi, K. et al. Le gène du récepteur de guidage axonal ROBO1 est un gène candidat pour la dyslexie développementale. PLoS Genet. 1 , e50 (2005).Article PubMed Centre PubMed Google Scholar
- Ganna, A. et al. Des mutations perturbatrices et dommageables ultra-rares influencent le niveau d’instruction dans la population générale. Nat. Neurosci. 19 , 1563-1565 (2016).Article CAS PubMed Centre PubMed Google Scholar
- Gialluisi, A. et al. Étude des effets des variantes du nombre de copies sur les performances en lecture et en langage. J. Neurodev. Désordre. 8 , 17–17 (2016).Article PubMed Centre PubMed Google Scholar
- Oksenberg, N., Stevison, L., Wall, JD & Ahituv, N. Fonction et régulation de AUTS2, un gène impliqué dans l’autisme et l’évolution humaine. PLoS Genet. 9 , e1003221 (2013).Article CAS PubMed Centre PubMed Google Scholar
- Beunders, G. et al. Deux hommes adultes avec des variantes pathogènes AUTS2, y compris une délétion de deux paires de bases, délimitent davantage le syndrome AUTS2. EUR. J. Human Genet. 23 , 803–807 (2015).Article CAS Google Scholar
- Girirajan, S. et al. Charge relative des grands CNV sur une gamme de phénotypes neurodéveloppementaux. PLoS Genet. 7 , e1002334 (2011).Article CAS PubMed Centre PubMed Google Scholar
- Wessel, K. et al. 17q23.2q23.3 duplication de novo associée à un trouble de la parole et du langage, des difficultés d’apprentissage, une incoordination, une déficience motrice et des troubles du comportement : à propos d’un cas. BMC Med. Genet. 18 , 119 (2017).Article PubMed Centre PubMed Google Scholar
- Guo, H. et al. Les mutations perturbatrices de TANC2 définissent un syndrome neurodéveloppemental associé à des troubles psychiatriques. Nat. Commun. 10 , 4679 (2019).Article PubMed Centre PubMed Google Scholar
- Pasmant, E. et al. Caractérisation d’une délétion germinale de 7,6 Mb englobant le locus NF1 et une centaine de gènes chez un patient atteint du syndrome des gènes contigus NF1. EUR. J. Hum. Genet. 16 , 1459-1466 (2008).Article CAS PubMed Google Scholar
- Takata, A. et al. Les analyses intégratives des mutations de novo fournissent des informations biologiques plus approfondies sur les troubles du spectre autistique. Cell Reports 22 , 734–747 (2018).Article CAS PubMed Google Scholar
- Lee, JJ et al. Découverte de gènes et prédiction polygénique à partir d’une étude d’association à l’échelle du génome sur le niveau de scolarité de 1,1 million d’individus. Nat. Genet. 50 , 1112-1121 (2018).Article CAS PubMed Centre PubMed Google Scholar
- Finucane, HK et al. Partitionnement de l’héritabilité par annotation fonctionnelle à l’aide de statistiques récapitulatives d’association à l’échelle du génome. Nat. Genet. 47 , 1228-1235 (2015).Article CAS PubMed Centre PubMed Google Scholar
- Grove, J. et al. Identification des variantes de risque génétique courantes pour les troubles du spectre autistique. Nat. Genet. 51 , 431–444 (2019).Article CAS PubMed Centre PubMed Google Scholar
- Mozzi, A. et al. L’histoire évolutive des gènes impliqués dans le langage parlé et écrit : au-delà de FOXP2. Sci. Rep. 6 , 22157 (2016).Article CAS PubMed Centre PubMed Google Scholar
- Schmitz, J., Abbondanza, F. & Paracchini, S. Étude d’association à l’échelle du génome et analyse du score de risque polygénique pour les mesures auditives chez les enfants. Un m. J. Med. Genet. B Neuropsychiatre. Genet. 186 , 318-328 (2021).Article CAS PubMed Google Scholar
- Vellutino, F. Conceptualisations alternatives de la dyslexie: preuves à l’appui d’une hypothèse de déficit verbal. Harvard Éduc. Rév. 47 , 334–354 (2012).Article Google Scholar
- Dehaene, S., Cohen, L., Morais, J. & Kolinsky, R. Illettré à alphabétisé : changements comportementaux et cérébraux induits par l’acquisition de la lecture. Nat. Rév. Neurosci. 16 , 234-244 (2015).Article CAS PubMed Google Scholar
- Cuellar-Partida, G. et al. Une étude d’association à l’échelle du génome identifie 48 variantes génétiques communes associées à la latéralité. Nat. Hum. Comportement 5 , 59–70 (2021).Article PubMed Google Scholar
- Papadatou-Pastou, M. et al. Human handedness: a meta-analysis. Psychol. Bull. 146, 481–524 (2020).Article PubMed Google Scholar
- Peters, M., Reimers, S. & Manning, JT Préférence manuelle pour l’écriture et associations avec des variables démographiques et comportementales sélectionnées chez 255 100 sujets: l’étude Internet de la BBC. Cerveau Cogn. 62 , 177-189 (2006).Article PubMed Google Scholar
- Brandler, WM & Paracchini, S. La relation génétique entre la latéralité et les troubles neurodéveloppementaux. Tendances Mol. Méd. 20 , 83–90 (2014).Article PubMed Centre PubMed Google Scholar
- Willcutt, EG, Pennington, BF, Olson, RK, Chhabildas, N. & Hulslander, J. Analyses neuropsychologiques de la comorbidité entre le trouble de lecture et le trouble déficitaire de l’attention avec hyperactivité : à la recherche du déficit commun. Dév. Neuropsychol. 27 , 35–78 (2005).Article PubMed Google Scholar
- Gu, X. et al. Réponse cérébrale accrue à l’anticipation de la douleur chez les adultes de haut niveau atteints de troubles du spectre autistique. EUR. J. Neurosci. 47 , 592–601 (2018).Article PubMed Google Scholar
- Whitney, DG & Shapiro, DN Prévalence nationale de la douleur chez les enfants et les adolescents atteints de troubles du spectre autistique. JAMA Pédiatre. 173 , 1203–1205 (2019).Article PubMed Centre PubMed Google Scholar
- Munafò, MR, Tilling, K., Taylor, AE, Evans, DM et Davey Smith, G. Portée du collisionneur : lorsque le biais de sélection peut influencer considérablement les associations observées. Int. J. Épidémiol. 47 , 226-235 (2018).Article PubMed Google Scholar
- Fontanillas, P. et al. Scores de risque de maladie pour les cancers de la peau. Nat. Commun. 12 , 160 (2021).Article CAS PubMed Centre PubMed Google Scholar
- Das, S. et al. Service et méthodes d’imputation de génotype de nouvelle génération. Nat. Genet. 48 , 1284-1287 (2016).Article CAS PubMed Centre PubMed Google Scholar
- Gialluisi, A. et al. L’analyse d’association à l’échelle du génome identifie de nouvelles variantes associées à un prédicteur cognitif de la dyslexie. Trad. Psychiatrie 9 , 77 (2019).Article PubMed Centre PubMed Google Scholar
- de Leeuw, CA, Mooij, JM, Heskes, T. & Posthuma, D. MAGMA : analyse généralisée des ensembles de gènes des données GWAS. Calcul PLoS. Biol. 11 , e1004219 (2015).Article PubMed Centre PubMed Google Scholar
- Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Cartographie fonctionnelle et annotation des associations génétiques avec FUMA. Nat. Commun. 8 , 1826 (2017).Article PubMed Centre PubMed Google Scholar
- Subramanian, A. et al. Analyse d’enrichissement d’ensembles de gènes : une approche basée sur les connaissances pour interpréter les profils d’expression à l’échelle du génome. Proc. Natl Acad. Sci. États-Unis 102 , 15545–15550 (2005).Article CAS PubMed Centre PubMed Google Scholar
- Liberzon, A. et al. Base de données de signatures moléculaires (MSigDB) 3.0. Bioinformatique 27 , 1739–1740 (2011).Article CAS PubMed Centre PubMed Google Scholar
- Wakefield, J. Une mesure bayésienne de la probabilité de fausse découverte dans les études d’épidémiologie génétique. Un m. J. Human Genet. 81 , 208–227 (2007).Article CAS Google Scholar
- Maller, JB et al. Raffinement bayésien des signaux d’association pour 14 loci dans 3 maladies courantes. Nat. Genet. 44 , 1294-1301 (2012).Article CAS PubMed Centre PubMed Google Scholar
- Howe, KL et al. Ensemble 2021. Nucleic Acids Res. 49 , D884–D891 (2020).Article Centre PubMed Google Scholar
- Carvalho-Silva, D. et al. Open Targets Platform : nouveaux développements et mises à jour deux ans après. Nucleic Acids Res. 47 , D1056–D1065 (2018).Article Centre PubMed Google Scholar
- Petrovski, S. et al. L’intolérance de la séquence régulatrice à la variation génétique prédit la sensibilité au dosage des gènes. PLoS Genet. 11 , e1005492 (2015).Article PubMed Centre PubMed Google Scholar
- Rada-Iglesias, A. H3K4me1 est-il corrélatif ou causal aux activateurs ? Nat. Genet. 50 , 4–5 (2018).Article CAS PubMed Google Scholar
- Heintzman, ND et al. Signatures chromatiniennes distinctes et prédictives des promoteurs et amplificateurs transcriptionnels du génome humain. Nat. Genet. 39 , 311–318 (2007).Article CAS PubMed Google Scholar
- Cahoy, JD et al. Une base de données de transcriptomes pour les astrocytes, les neurones et les oligodendrocytes : une nouvelle ressource pour comprendre le développement et le fonctionnement du cerveau. J. Neurosci. 28 , 264 (2008).Article CAS PubMed Centre PubMed Google Scholar
- Le Consortium GTEx. Analyse pilote de l’expression génotype-tissu (GTEx) : régulation des gènes multitissulaires chez l’homme. Sciences 348 , 648 (2015).Article Google Scholar
- Finucane, HK et al. L’enrichissement de l’héritabilité des gènes spécifiquement exprimés identifie les tissus et les types de cellules pertinents pour la maladie. Nat. Genet. 50 , 621–629 (2018).Article CAS PubMed Centre PubMed Google Scholar
- Dunham, I. et al. Une encyclopédie intégrée des éléments d’ADN dans le génome humain. Nature 489 , 57–74 (2012).Article CAS Google Scholar
- Vermunt, MW et al. Annotation épigénomique des altérations régulatrices des gènes au cours de l’évolution du cerveau des primates. Nat. Neurosci. 19 , 494-503 (2016).Article CAS PubMed Google Scholar
- Reilly, SK et al. Génomique évolutive. Changements évolutifs dans l’activité du promoteur et de l’amplificateur au cours de la corticogenèse humaine. Sciences 347 , 1155-1159 (2015).Article CAS PubMed Centre PubMed Google Scholar
- Peyrégne, S., Boyle, MJ, Dannemann, M. & Prüfer, K. Détection d’une sélection positive ancienne chez l’homme à l’aide d’un tri de lignée étendu. Génome Res. 27 , 1563-1572 (2017).Article PubMed Centre PubMed Google Scholar
- Simonti, CN et al. L’héritage phénotypique du mélange entre les humains modernes et les Néandertaliens. Sciences 351 , 737-741 (2016).Article CAS PubMed Centre PubMed Google Scholar
- Vernot, B. et al. Excavation de l’ADN néandertalien et dénisovien à partir des génomes d’individus mélanésiens. Sciences 352 , 235-239 (2016).Article CAS PubMed Centre PubMed Google Scholar
- Gazal, S. et al. L’architecture dépendante du déséquilibre de liaison des traits complexes humains montre l’action de la sélection négative. Nat. Genet. 49 , 1421–1427 (2017).Article CAS PubMed Centre PubMed Google Scholar
- Bulik-Sullivan, BK et al. La régression du score LD distingue la confusion de la polygénicité dans les études d’association à l’échelle du génome. Nat. Genet. 47 , 291–295 (2015).Article CAS PubMed Centre PubMed Google Scholar
- Bulik-Sullivan, BK et al. Un atlas des corrélations génétiques entre les maladies et les traits humains. Nat. Genet. 47 , 1236-1241 (2015).Article CAS PubMed Centre PubMed Google Scholar
- Zheng, J. et al. LD Hub : une base de données centralisée et une interface Web pour effectuer une régression du score LD qui maximise le potentiel des données GWAS de niveau sommaire pour l’héritabilité des SNP et l’analyse de corrélation génétique. Bioinformatique 33 , 272–279 (2016).Article PubMed Centre PubMed Google Scholar
- Grasby, KL et al. L’architecture génétique du cortex cérébral humain. Sciences 367 , eaay6690 (2020).Article CAS PubMed Centre PubMed Google Scholar
- Satizabal, CL et al. Architecture génétique des structures cérébrales sous-corticales chez 38 851 individus. Nat. Genet. 51 , 1624-1636 (2019).Article CAS PubMed Centre PubMed Google Scholar
- Hibar, DP et al. Nouveaux locus génétiques associés au volume de l’hippocampe. Nat. Commun. 8 , 13624 (2017).Article CAS PubMed Centre PubMed Google Scholar
- Adams, HH et al. Nouveaux locus génétiques sous-jacents au volume intracrânien humain identifiés par association à l’échelle du génome. Nat. Neurosci. 19 , 1569-1582 (2016).Article CAS PubMed Centre PubMed Google Scholar
- Smith, SM et al. Un ensemble élargi d’études d’association à l’échelle du génome des phénotypes d’imagerie cérébrale dans UK Biobank. Nat. Neurosci. 24 , 737–745 (2021).Article CAS PubMed Centre PubMed Google Scholar
- Alfaro-Almagro, F. et al. Traitement d’image et contrôle qualité pour les 10 000 premiers ensembles de données d’imagerie cérébrale de UK Biobank. Neuroimage 166 , 400–424 (2018).Article PubMed Google Scholar
- Forkel, SJ & Catani, M. Le manuel d’Oxford de neurolinguistique : méthodes d’imagerie de diffusion en sciences du langage (Oxford Univ. Press, Oxford, 2019).Google Scholar
- Price, CJ L’anatomie du langage : une revue de 100 études IRMf publiées en 2009. Ann. NY Acad. Sci. 1191 , 62–88 (2010).Article PubMed Google Scholar
- Richardson, FM & Price, CJ Études structurelles par IRM de la fonction du langage dans le cerveau non endommagé. Structure du cerveau. Fonct. 213 , 511–523 (2009).Article PubMed Centre PubMed Google Scholar
- Perdue, MV, Mednick, J., Pugh, KR et Landi, N. La structure de la matière grise est associée à la compétence en lecture chez les jeunes lecteurs en développement typique. Cereb. Cortex 30 , 5449–5459 (2020).Article PubMed Centre PubMed Google Scholar
- Roehrich-Gascon, D., Small, SL et Tremblay, P. Corrélats structurels des capacités du langage parlé : une étude de morphométrie de la région d’intérêt basée sur la surface. Cerveau Lang. 149 , 46–54 (2015).Article PubMed Centre PubMed Google Scholar
- Luciano, M. et al. Une étude d’association à l’échelle du génome pour les capacités de lecture et de langage dans deux cohortes de population. Gènes Brain Behav. 12 , 645–652 (2013).Article CAS PubMed Centre PubMed Google Scholar
- Gialluisi, A. et al. Dépistage à l’échelle du génome des variants d’ADN associés aux traits de lecture et de langage. Gènes Brain Behav. 13 , 686–701 (2014).Article CAS PubMed Centre PubMed Google Scholar
- Purcell, S. et al. PLINK : un ensemble d’outils pour l’association du génome entier et les analyses de liaison basées sur la population. Un m. J. Human Genet. 81 , 559-575 (2007).Article CAS Google Scholar
- Euesden, J., Lewis, CM & O’Reilly, PF PRSice : logiciel de score de risque polygénique. Bioinformatique 31 , 1466–1468 (2015).Article CAS PubMed Google Scholar
- Bates, TC et al. Analyses génétiques comportementales de la lecture et de l’orthographe : une approche par processus composants. Aust. J. Psychol. 56 , 115–126 (2004).Article Google Scholar
- Dollaghan, C. & Campbell, TF Répétition de non-mots et troubles du langage chez l’enfant. J. Discours Lang. Écouter. Rés. 41 , 1136-1146 (1998).Article CAS PubMed Google Scholar
- Gathercole, SE, Willis, CS, Baddeley, AD & Emslie, H. Le test de répétition des non-mots pour enfants : un test de mémoire de travail phonologique. Mémoire 2 , 103–127 (1994).Article CAS PubMed Google Scholar
- L’équipe RStudio. RStudio : Développement intégré pour R. (Boston, MA, 2020).
- Ayub, Q. et al. Les cibles FOXP2 montrent des preuves de sélection positive dans les populations européennes. Un m. J. Human Genet. 92 , 696-706 (2013).Article CAS Google Scholar
- Poelmans, G., Buitelaar, JK, Pauls, DL et Franke, B. Un réseau moléculaire théorique pour la dyslexie : intégrer les découvertes génétiques disponibles. Mol. Psychiatrie 16 , 365–382 (2011).Article CAS PubMed Google Scholar
- Guidi, LG et al. L’hypothèse de la migration neuronale de la dyslexie : une évaluation critique 30 ans après. EUR. J. Neurosci. 48 , 3212–3233 (2018).Article PubMed Centre PubMed Google Scholar



